This condition is now divided
into ante-natal, intrapartum and post-natal, though
these are not necessarily mutually exclusive. Indeed
Stanley ( 1)
suggests that a cluster of intrapartum events contributing
to causal pathways can be found in 16% of children,
but only in 10% is there no evidence of additional
antenatal damage. Intrapartum and postnatal causes
will not be considered further in this review. 1)
suggests that a cluster of intrapartum events contributing
to causal pathways can be found in 16% of children,
but only in 10% is there no evidence of additional
antenatal damage. Intrapartum and postnatal causes
will not be considered further in this review.
ANTENATAL CAUSES
Genetic causes have been recognised in a few
families, but no specific inheritance pattern has been
identified, and inheritance appears to be multifactorial.
Rates of fetal and neonatal loss are higher in families
with all types of CP, as is a family history of being
small for dates. It is unclear what the relative contribution
of genetic, environmental and nutritional factors is
in most families.
Congenital malformations have a strong association
with CP. This suggests that damage to the fetus' neurological
system is early in the pregnancy, rather than later.
Data collected in Western Australia on CP showed 29%
of those children with moderate or severe CP had a
congenital malformation compared to 4.9% for the control
group and normal population (2, 3).
CP is also known to be more common in boys than girls.
Multiple pregnancies carry an increased risk
of CP as an outcome. In a recent meta-analysis, Stanley
calculated a relative risk for CP (compared to singletons)
of 4.5 (95% CI 3.9, 5.2) for twins and 18.2 (10.4,
31.9) for triplets (4).
The risk remains after correction for increased rates
of prematurity. The risk is particularly high when
one twin or triplet dies. The relative risk for a twin
whose co-twin died is 11.4 (7.0, 18.8) compared to
twins where both survive, and this is a 40 fold increase
compared to the singleton population (3).
This has led Pharoah (5)
to postulate that 'the disappearing twin' - i.e. very
early loss of a twin fetus - may be the cause of CP
in a number of (apparent) singletons (5).
| Maternal infections are well-recognised
causal pathways of CP. Historically the TORCH infections
were first described. However, congenital rubella
is relatively rarely seen in the UK due to screening
and childhood vaccinations. CMV (cytomegalovirus)
infection is estimated to affect around 0.3% of
fetuses in utero (6),
damage being more severe if in the first or second
trimester of pregnancy. |
|
TOxoplasmosis
Rubella
Cytomegalovirus
Herpes simplex |
|
More recently, interest has been directed at other
bacterial and mycoplasmal infections (chorioamnionitis,
bacterial vaginosis, urinary tract infections) and
whether they result in premature labour - the latter
a risk factor for CP.
A study from California showed an association between
CP and infection in term babies weighing over 2500gms.
A fever of over 38 degrees in labour was associated
with a 9-fold risk of CP overall and a 19 fold increase
in quadriplegia. Chorioamnionitis specifically had
a risk of 4.2 to 12 fold of CP, and up to 31-fold for
quadriplegia (7).
The group investigated this by looking for inflammatory
markers in the neonatal blood spots of a group of CP
children more than 3 years old, and comparing their
concentrations with those found in the neonatal blood
spots of a group of control children. A complex picture
emerged, but there were significantly raised concentrations
of two groups of chemicals in the blood spots of the
CP children (8).
65% of the CP children had raised autoimmune antibodies
and/or abnormal coagulation factors, compared to 3%
of the controls. As a group they also had raised levels
of a number of cytokines, including IL-1, IL-6, IL-8
and Tumour Necrosis Factor alpha (TNF). The inflammatory
markers were thought to be secondary to the acute infections.
The coagulation factors pointed, perhaps separately,
to the significance of coagulopathy and interruption
of cerebral blood supply - even at a microvascular
level - as pathways to brain damage and consequent
CP. Studies showing a relationship between peri- and
post-natal infections have now been reported from the
UK (9, 10).
One theoretical model to link this findings as cause
and effect was proposed by Leviton in 1990 (11).
He proposed that chorioamnionitis and/or maternal or
uterine infections caused a release of TNF, either
directly or as a result of endotoxin release. TNF might
then stimulate prostaglandin release and cause preterm
labour, and the preterm baby would then face a series
of hazards (particularly hypotension and hypoxia) that
could lead to brain damage. Alternatively the TNF might
target specific areas of the brain and cause a localised
inflammatory response resulting in permanent damage.
Infection and preterm labour
There are reported associations between bacterial vaginosis
(12),
urinary tract infections and asymptomatic bacteriuria
(13)
and preterm labour. The significance of these in relation
to CP needs elucidation. CP is increased in preterm babies
(14),
but is unclear whether the causative pathway is through
the complications of prematurity alone, or whether cytokines
cause neuro-chemical damage as appears to be the case
with chorioamnionitis and periventricular leucomalacia
(see below)
Treatment of asymptomatic bacteriuria probably prolongs
pregnancy (15),
and treatment of women with bacterial vaginosis and
a previous preterm birth reduces the number of preterm
deliveries (16).
The current Cochrane review of the treatment of PROM
with antibiotics does not advocate 'routine' treatment
with antibiotics as there is no clear benefit for overall
neonatal morbidity or mortality, although fewer babies
need prolonged intensive care (17).
However neither this nor the other reviews include
good quality data on the long-term neurological outcome
for the neonates. The results of the UK ORACLE study,
coordinated by the author of the review on PROM, are
still awaited. However, no long term benefit is recognised
in giving antibiotics to women in preterm labour with
intact membranes, although there is some reduction
in NEC (18,19).
Infection and Periventricular leucomalacia
Support for the concept that chorioamnionitis might
cause brain damage in the fetus came from studies in
America and France in 1996. Perlman's team in Dallas
identified bilateral cystic periventricular leucomalacia
(CVPL) in 14 (2.3%) of 632 infants weighing less than
1750g at birth. Only 4 of these had ever been hypotensive,
which was previously thought to be a major pathway
to PVL, though other studies had not reported this
consistently. They examined 20 perinatal risk factors
for morbidity and univariate analysis showed that only
chorioamnionitis and prolonged rupture of the membranes
(PROM) were significantly associated with PVL (20).
For PROM the OR was 6.6 (95% CI 2, 22.1) and for chorioamnionitis
6.8(1.8, 25.9). Similar results came from France (21).
Although not within the scope of this review, it is
important to remember that post-natal hypotension can
lead to PVL, and Grade 4 IVH or periventricular venous
haemorrhagic infarction cause white matter damage with
a high risk of neurological impairment in survivors.
| Periventricular leukomalacia is 'softening of
the white matter around the ventricles'. Cystic
periventricular leukomalacia results from extensive
necrosis of white matter adjacent to lateral ventricles
in the brain. This area includes the cortico-spinal
(pyramidal) tracts, and the optic radiations, so
neurological consequences include motor deficits
such as diplegia, and cortical blindness.
Initial studies suggested hypoxia and underperfusion
as risk factors associated with PVL, and it
was therefore ischaemic in origin. PVL is seen
after neonatal hypotension (mean BP < 30mmHg
in the early neonatal period), but not consistently
so, and not all studies have found hypotension
to be a significant association. Recently neuro-chemically
mediated injury has been recognised as a potential
mechanism for white matter damage.
Normally it is detected by ultrasound scans
of the brains of infants undergoing intensive
care. It shows as an echodensity that may or
may not progress to cystic changes. Autopsy
findings in infants who die with PVL show cell
swelling and death of brain cells, with acute
inflammatory changes. After death of the neural
cells areas of gliosis (mild cases) or cyst
formation (severe cases) develop. These may
occur at the microscopic level with no obvious
scan findings in the mild cases. If cysts develop,
they become apparent from about 12 days after
the causative insult. Thus a baby who develops
cystic PVL in the first week of life has suffered
an antenatal insult.
The ultrasound findings are graded into:
- Flare
- Persistent flare (> 14 days)
- Cystic PVL
These changes may be bilateral or unilateral
These have some prognostic significance. In
one study all 14 babies with cysts > 5mm
developed CP, as did 2/11 babies with smaller
cysts and 6/12 babies with prolonged flare
(22).
Other studies have reported similar but not
identical outcomes with the size and site of
the cysts being important.
|
|
|
|
| |
|
|
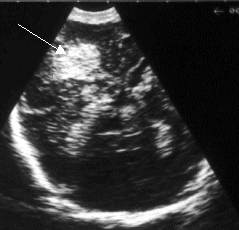
| Figure 1a: A preterm baby with
a left sided flare. It shows the typical
triangular shape flaring out from the lateral
tip of the lateral ventricle. |
|
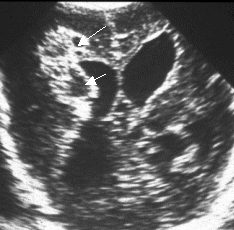
Figure 1b: The same baby 3 weeks
later with extensive cystic changes (arrowed)
within the 'flare': this is cystic PVL.
The ventricles are now dilated. |
| |
|
|
|
|
|
|
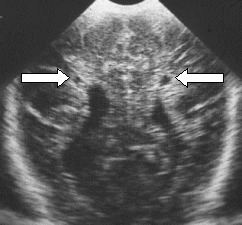
| Figure 2: A baby with bilateral
cystic changes (arrowed) (see reference
to Perlman et al in show main text) |
|
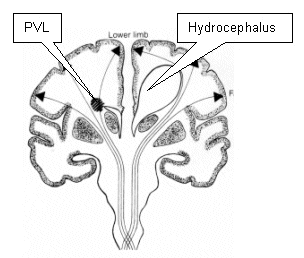
Figure 3 A simplified diagram of the
cortico-spinal tracts, to et al in et how
PVL and hydrocephalus may affect them. |
| |
|
|
|
Deficiencies of thyroid and iodine in the mother
can have a profound effect on the fetus. The fetus
can not produce its own thyroid hormone until late
in the first trimester and relies on maternal T4 crossing
the placenta; T4 being important
for neuronal development. Iodine deficiency is linked
with a spectrum of motor disorders.
Fetal growth
Growth restriction is one of the most important risk
factors associated with CP, although the association
appears more significant for term infants than for
premature infants (23,24,25).
This may be because the term infants have been under
stress for a longer period of time or because the premature
symmetrically growth restricted babies have poor survival
rates and are therefore not reflected in the statistics.
This link with CP may be that growth restricted babies
are more vulnerable to insult, particularly intrapartum
stress (24).
What is not clear is whether CP causes the baby to
grow poorly or whether IUGR makes the baby more susceptible
to cerebral damage and CP.
Pre-eclampsia and Preventative Treatment
PET itself is associated with a decreased risk of
CP, independent of magnesium sulphate usage (25).
It is associated with decreased IVH (intraventricular
haemorrhage) in the preterm infant and a decrease in
infection (25)
that might help explain this. Equally, there is a high
incidence of elective delivery in this group (23)
Mode of delivery, or more specifically, delivery without
labour appears to have a protective effect against
neurological damage. Antenatal steroids reduce IVH,
RDS ( Respiratory distress syndrome) and necrotising
enterocolitis (26).
Steroids are given when there is a risk of premature
delivery, and may be another confounding variable in
assessing the effect of PET.
Retrospective and observational studies suggest that
magnesium sulphate, when used for eclampsia or as a
tocolytic, may have a protective effect against CP
(9),
with 3 possible mechanisms:
- Reversing cerebral arterial vasoconstriction of
middle cerebral artery
- Releasing endothelial prostacyclins
- Inhibiting platelet aggregation
A MAGnet trial (Magnesium and neurological endpoints
trial) was therefore set up as a double-blind RCT,
comparing magnesium with a placebo. PET was excluded
as a confounding variable. Unfortunately there were
significantly higher deaths reported in the magnesium
arm and the trial had to be stopped prematurely (27).
References
1. Stanley F, Blair E, Alberman E. Pathways to cerebral
palsy involving signs of birth asphyxia. In: Cerebral
Palsies: Epidemiology and Causal Pathways. Clin Devlop
Med 2000;151:22-39.
2. Stanley FJ. Survival and cerebral palsy in low
birthweight infants: implications for perinatal care.
Paediatric and Perinatal Epidemiology 1992;6:298-310, Abstract
3. Palmer L. Antenatal antecedents of moderate and
severe cerebral palsy. Paediatric and Perinatal Epidemiology
1993;16:95-102, Abstract
4. Stanley F, Blair E, Alberman E. The Special case
of Multiple Pregnancy? In: Cerebral Palsies: Epidemiology
and Causal Pathways. Clin Devlop Med 2000;151:109-123
5. Pharoah PO, Adi Y. Consequences of in-utero death
in a twin pregnancy. Lancet 2000;355:1597-1622, Abstract
6. Logan S: personal communication 1998.
7. Grether JK, Nelson K Maternal infection and cerebral
palsy in infants of normal birth weight. JAMA;1997;278:207-11, Abstract
8. Nelson K et al. Neonatal Cytokines and Coagulation
in Children with CP. Ann Neurol 1998;44:665-675, Abstract
9. Murphy D. Case-control study of antenatal and intrapartum
risk factors for cerebral palsy in very preterm singleton
babies. Lancet 1995;346:1449-54, Abstract
10. Wheater M. Rennie JM. Perinatal infection is an
important risk factor for cerebral palsy in very-low-birthweight
infants. Developmental Medicine & Child Neurology.
2000; 42:364-7, Abstract
11. Leviton A, Paneth N White matter damage in preterm
newborns: an epidemiologic perspective. Early Human
Development 1990;24:1-22, Abstract
12. Hay PE, Lamont RF, Taylor-Robinson D, Morgan DJ,
Ison C, Pearson J. Abnormal bacterial colonisation
of the genital tract and subsequent preterm delivery
and late miscarriage. BmedJ 1994;308:295-298, Abstract
13. Moller M, Thomsen AC, Borch K, Dinesen K, Zdravkovic
M. Rupture of fetal membranes and premature delivery
associated with Group B Streptococci in urine of pregnant
women. Lancet 1984;Iii;69-70, Abstract
14. Stanley F, Blair E, Alberman E. Pathways to cerebral
palsy involving preterm birth. In: Cerebral Palsies:
Epidemiology and Causal Pathways. Clin Dev Med 2000;151:109-123, Abstract
15. Smaill F. Antibiotics for asymptomatic bacteriuria
in pregnancy. Cochrane Database of Systematic Reviews,
2000.Abstract
16. Brocklehurst P, Hannah M, McDonald H. Interventions
for treating bacterial vaginosis in pregnancy. Cochrane
Database of Systematic Reviews, 2000, Abstract
17. Kenyon S. Antibiotics for preterm premature rupture
of the membranes. Cochrane Database of Systematic Reviews,
2000, Abstract
18. King J. Antibiotics for preterm labour with intact
membranes. Cochrane Database of Systematic Reviews,
2000, Abstract
19. Norman K, Pattinson RC, deSouza J, de Jong P,
Moller G, Kirsten G. Ampicillin and Metronidazole treatment
in preterm labour: a multicentre randomised controlled
trial. Br J Obst Gynae 1994;101:404-8, Abstract
20. Perlmann JM, Risser R, Broyles RS. Bilateral cystic
Periventricular Leukomalacia in the Premature Infant:
Associated Risk Factors. Pediatr 1996;97:822-27,Abstract
21. Zupan V, Gonzalez P, Lacaze-Masmonteil et al.
Periventricular leucomalacia: risk factors revisited.
Dev Med Child Neurol 1996;38:1061-67, Abstract
22. Fazzi E, Orcesi S, Caffi L, Ometto A, Rondini
G, Telesca C, Lanzi G. Neurodevelopmental outcome at
5-7 years in preterm infants with periventricular leukomalacia.
Neuropediatrics. 1994;25:134-9.
23. Blair E. Intrauterine growth and spastic cerebral
palsy 1: association with birth weight for gestational
age. Am J Obstet Gynecol 1990;162:229-37,Abstract
24. Berg AT. Childhood neurological morbidity and
its association with gestational age, intrauterine
growth retardation and perinatal stress, Abstract
25. O'Shea TM. Antecedents of cerebral palsy in very
low-birth weight infants. Clinics in Perinatology 2000;27(2):285-302, Abstract
26. Crowley P. Antenatal corticosteroid therapy: a
meta-analysis of the randomised trials, 1972 to 1994.
Am J Obstet Gynecol 1995;173:322-35, Abstract
27. Mittendorf R. Is tocolytic magnesium sulphate
associated with increased total paediatric mortality?
Lancet 1997;350(9090), Abstract
28. Spinillo A. Obstetric risk factors for periventricular
leucomalacia among preterm infants. Br J Obstet Gynaecol
1998;105:865-71, Abstract
|